01. 생물정보학 주요 분야
1) 유전체학(Genomics)
- 개체의 전체 유전 정보 연구
- DNA염기 서열 및 유전자 발현을 분석하여 유전자의 기능과 조절 연구
2) 단백체학(Proteomics)
- 전체 단백질의 조성 / 기능 / 단백질 간의 상호작용, 단백질의 조절 연구
3) 시스템생물학(Systems biology)
- 생물학적 시스템을 전체적으로 이해
- 여러 생물학적 구성 요소간의 상호작용 및 네트워크 연구
4) 구조생물학(Structural biology)
- 단백질간의 구조 (ex. 어떤 부분 / 구조에서 상호작용되는지 보는 것)
5) 진화생물학(Evolutionary biology)
- 생물 다양성과 진화 과정을 연구하여 생물의 종류와 형태의 변화를 이해하고 설명
02. Next Generation Sequencing (NGS)
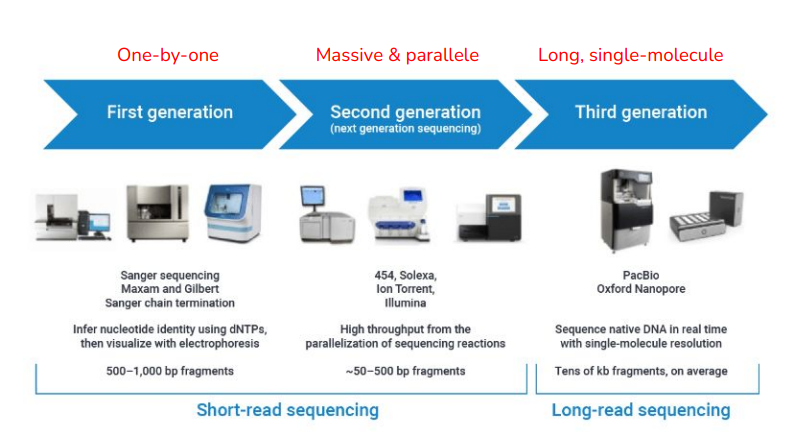
< Short-read sequencing >
1) First Generation
- 한 번에 하나의 시퀀스를 읽음(one-by-one)
- Sanger sequencing(생어 시퀀싱)
- 500~1000bp fragments(500~1000개의 조각)
2) Second generaion
- Next Generation Sequencing
- 대량 & 동시에 병렬적으로 데이터를 생산해 내는 기술
- illumina sequencing(일루미나 시퀀싱)
- ~50- 500 bp fragments (50~500개의 조각)
< Long-read sequencing >
3) Third generation
- 유전자를 쪼개지 않고 통째로 시퀀싱
- 바로 서열을 결정
- Tens of kb fragments, on average
03. NGS workflow
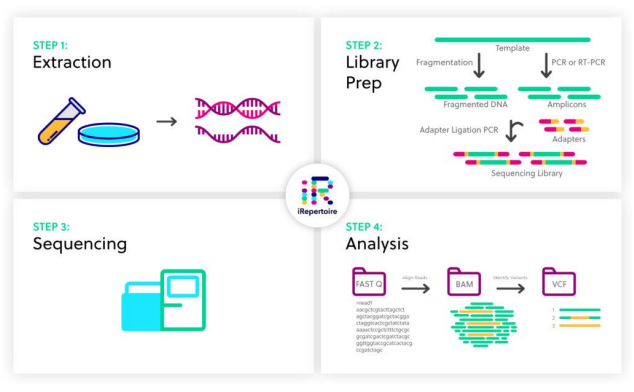
1) Extraction(샘플 추출)
- DNA or RNA 선택해서 샘플 추출
- 무엇을 분석할지 정하는 단계
- 즉, NGS를 어디에 적용할지 정하는 단계
2) Library prep
3) Sequencing
4) Analysis
3-1) Library prep(Library Preparation)

- DNA 또는 RNA 중, 무엇을 가지고 분석하고 싶은지 결정한 뒤, Library(라이브러리)를 만드는 작업
- DNA ploishing : Input DNA/RNA를 쪼갬 -> 쪼개고 나면, 보통 끝부분이 지저분하게 잘리기 때문에 끝부분을 다듬는 /복구하는 과정 진행
- Adaptor Ligation: adaptor 붙임

* p5 /p7 : flow cell에 binging 하는 adaptor
* Index1 /2 : 샘플의 바코드 역할 / normal과 암을 동시에 장비에 넣어서 샘플을 만들 때(비용을 아끼기 위해), 각 샘플이 어디에서 왔는지 알아보기 위함
* Rd 1/2 Sp : 실질적으로 리드가 읽히기 시작되는 부분 / DNA insert가 붙는 부분
- Size selection : 이렇게 만들어진 것들은 사이즈가 제각각일 텐데, 특정 범위에 있는 리드들만 가져옴
3-2) Sequencing
- DNA에서 뉴클레오타이드의 순서인 염기서열을 결정하는 과정
*뉴클레오티드 염기 : 아데닌(A), 시토신(C), 구아닌(G), 티민(T)
- Sample loading onto a flow cell
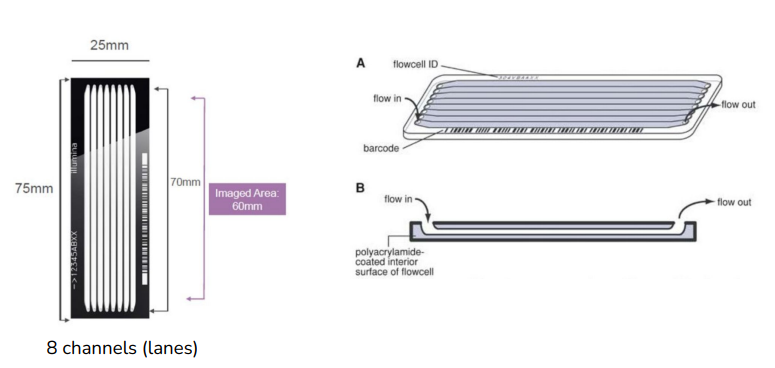
flow cell('판'이라고 생각하면 됨)에 샘플들을 올림. 총 8개의 채널이 있고, 한 채널당 만들 수 있는 데이터의 양이 정해져 있음. 한 채널에 여러 개의 심플을 올림(multiplexing기술)
- Sequencing DNA fragments
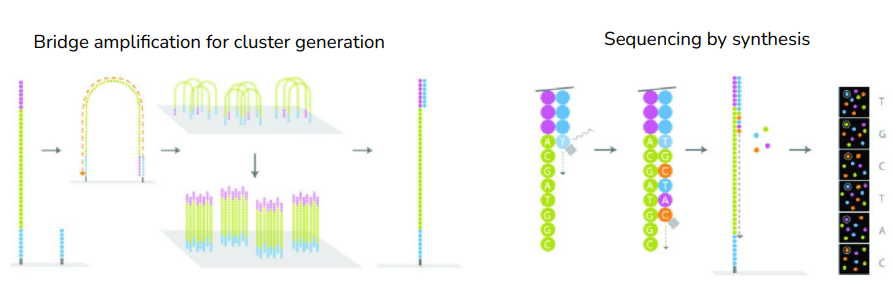
-> Bridge amplification for cluster generation : 동일한 리드에서 생성된 클러스터들이 생김. 각 클러스터마다 같은 시퀀스를 가짐. 이렇게 클러스터가 형성되어야 시그널을 조금 더 명확하게 읽어낼 수 있음(하나보다 여러 개가 뭉쳐있으면 더 신호가 커지니까)
-> Sequencing by synthesis : 실제로 시퀀스를 읽는 작업. 합성해 나가면서 염기서열을 결정하는 것.
=> 이러한 과정을 통해 'fastq file'을 얻을 수 있다.
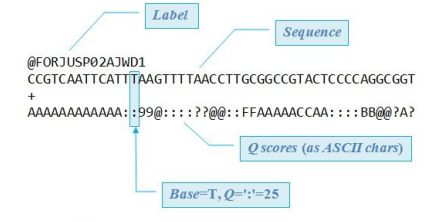
**fastq file
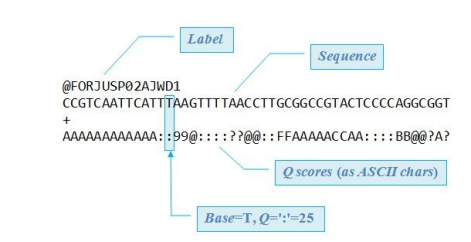

-fastq file에는 각각 어떤 염기 서열을 가지는지, 리드 identify를 가지는지 등을 알 수 있음
- 특히, Q-scores를 보면 얼마나 정확히 염기가 결정되어 있는지 (서열에 대한 퀄리티 정보)를 알 수 있음
- 예를 들어, Q30이면, 1 in 1000(1000개 중, 한 개의 에러가 나오는 수준)이라는 의미이며, 보통 Q30을 glodstandard로 설정함.
- Q-score에서 Q값이 높을수록 정확도가 높은 것
04. NGS data preprocessing workflow
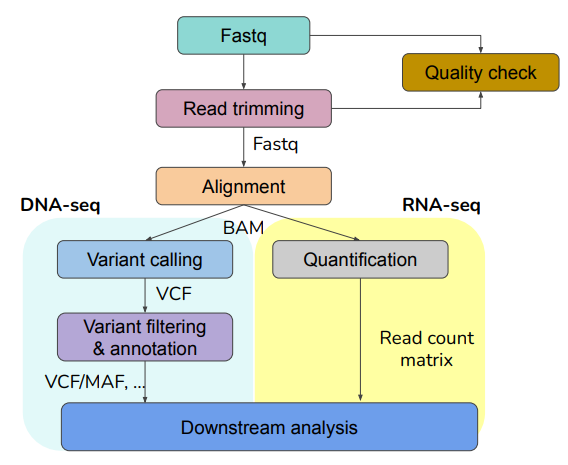
이전의 과정에서 만들어진 fastq file을 이용하며, 어떻게 분석하는지에 대한 workflow.
fastq file(raw data)만 가지고는 한 번에 무언가를 알아내기 힘들기 때문에 추가로 위와 같은 작업을 거침.
4-1) Quality check (QC)
- Fastq file과 Read trimming(약간 거르고) 후, Fastq file을 비교해서 Quality check 해봄 (리드 서열들이 얼마나 깔끔하게 읽혔는지)
- fastqc(가장 대표적인 방법)
4-2) Trimming
- 퀄리티가 안 좋은 서열을 잘라버리는(거르는) 작업
- Trimming 하는 이유
1) 리드에 포함된 adaptor 시퀀스를 제거하기 위함
(만약 fragment의 길이가 우리가 읽어내는 read의 길이보다 짧으면, adapter 부분까지 쭉 넘어 포함해서 읽어버리게 됨. 따라서 이러한 경우 adapter 부분을 자르는 과정이 필요한 것)
2) 말단에서 퀄리티 낮은 부분들(Q20 이하)을 제거
4-3) Alignment
- Trimming을해서 깔끔한 fastq file을 얻고 난 후, 각 리드들이 어디에서 왔는지 알기 위해 이를 레퍼런스 genome에 맵핑(레퍼런스 시퀀스와 내가 추출한 fastq file을 대조해 보는 것)
- DNA와 RNA에서 alignment방법이 다름
- fastq file을 Alignment 진행(레퍼런스 맵핑)하면 'BAM file'이 생성됨
4-4) Variant Calling
- BAM file로부터 유전적 변이를 calling 하는 단계(변이를 찾는 단계)
- 이 과정으로부터 'VCF(variant calling file)'이 생성됨
- VCF에는 어떤 변이가 이 환자에게 있는지 총체적 정보가 담겨있으며, 어떤 calling프로그램을 썼는지에 따라서 출력 정보가 달라짐
** fastq file vs BAM file vs Variant calling file(VCF)
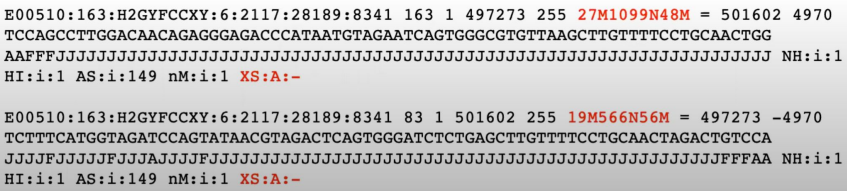

4-5) Variant Annotation(DNA-seq)
- 각 변이들을 annotation(이 변이가 질병과 관련해서 의미가 있는 것인지 분석)해주는 작업
'🩺 의학' 카테고리의 다른 글
| [의학] 임상 의학용어(Cardiovascular disease 위주) (0) | 2024.06.02 |
|---|---|
| [유전체 분석] 3. Proteomics / Protein analysis (1) | 2023.10.30 |
| [유전체 분석] 2. RNA-sequencing (0) | 2023.10.29 |

